This is a re-up of something I posted on my other blog, but it seemed fitting to move it over to share here also.
____________________________
I happen to be "reminiscing" about the good ol' days with my pal & decided to peruse my old myspace account (that I havent touched in well over a year!) & came across the blog post I did back in 2007 when Mailyna was granted "her wish" to go to Disney World her kindergarten year. It made me remember her kindergarten year today when I ran into another parent at the "ice cream social" at Mailyna's school & we chatted about our kids being in the same classroom this year & remembering when they were little kinderbabies. So heres the article that from 2007, that ran my baby on the front page of the Daily Review.
______________________________
Straight off Myspace:
Below is the article ran in Sept 8th's Daily Review about Mailyna. Make a Wish is real & does great things for kids who truely deserve a little bit of sunshine in their lives. I'll be forever greatful that my daughter was giving such a wonderful opportunity...
WISH COMES TRUE
Fairview girl gets trip to Disney
Foundation provides vacation for 4-year-old suffering from disease
Article Last Updated: 09/08/2007 09:28:16 AM PDT

Almia Armas and her daughter Mailyna Mayate, 4, in their Hayward home Thurs. Sept. 6, 2006, are going to Walt Disney World in Florida with the help of Make A Wish foundation. Mailyna suffers from a life-threatening blood disease.
FAIRVIEW — Mailyna Mayate resembles a little princess in her pink-spangled gown, rhinestone tiara and wand sparkling with pixie dust.
The 4-year-old feels like one, too.
In late August, she started kindergarten at Stanton Elementary School in Castro Valley. On Sept. 16, she turns 5. And, she was asked Thursday, "Mailyna, what are you doing Saturday?"
"I'm going to Disney World," she said, giggling before collapsing in a fluffy heap to work on Cinderella and Little Mermaid puzzles.
Yes, at 11 a.m. today, a lim-ousine is taking Mailyna, her mother and her grandmother to San Francisco International Airport.
They'll fly to Florida and stay five nights at "Give Kids the World" Village, a resort in Kissimmee, where Mailyna will eat at the Gingerbread House. They'll also have passes to four Disney theme parks, Universal Studios and Sea World in neighboring Orlando.
A special trip for a special little girl?
Yes, and the fulfillment of a special wish by the Greater Bay Area Make-A-Wish Foundation for a child who has undergone more physical turmoil in her short life than many adults.
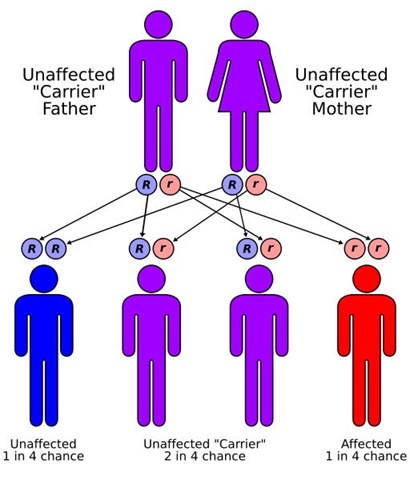
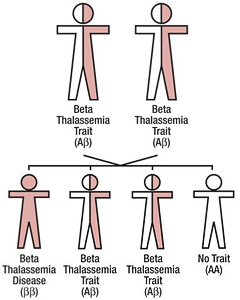
Mailyna suffers from thalassemia, a life-threatening blood disease. Her body cannot produce red blood cells.
She has had blood transfusions every five weeks since she was 6 months old. Just before her first birthday, a port — or small reservoir — was implanted in her chest for the transfusions. It lasts five to seven years before a replacement is necessary.
If her body temperature exceeds 101.5 degrees, Mailyna is hospitalized for three days for antibiotic therapy. She has liver biopsies yearly. She's currently not a candidate for a bone marrow transplant, which could introduce blood-forming cells into her body.
In addition, Mailyna takes medication every day to reduce the effects of iron overloads on her body caused by the transfusions. The oral medicine is a blessing, said Mailyna's mother, Almia Armas.
Until a year ago, Armas had to prick Mailyna's arms or legs with a syringe at bedtime, so that medication could pump through her daughter's body overnight. The child's limbs grew hard and callused, and it hurt her to walk.
Now, looking at affectionate, effervescent Mailyna, you would never know she is anything but a normal child.
"She is normal," Armas insisted. "She plays, swims and dances Tahitian dances. She loves to go camping and to the snow, to work on the computer and to take pictures with my camera."
In part, Mailyna accepts her illness "because she knows nothing else," explained her grandmother, Eileen Armas.
Love and diligence account for Mailyna's quality of life.
She lives with her mother, grandparents, aunt and uncle near Don Castro Regional Recreation Area. Mailyna's father is not involved in her life, Almia Armas said.
The family has good medical coverage, in part due to Armas's employment and insurance plan at Kaiser Medical Center in Oakland, where Mailyna is treated in the pediatric hematology department.
"I wouldn't trade my life for anything else," Almia Armas, 25, said as Mailyna cried "Mommy!" and leaped into her arms. "She's my world."
And, for five days, Mailyna's world will be one of princesses and heroines whose names she reels off affectionately and quickly:
"Aurora (Mailyna's middle name). Cinderella. Sleeping Beauty. Snow White. Ariel. Belle. The Little Mermaid."
_____
Personal Disclaimer: I am sorry that in the paper they printed my statement that her father is not involved... while he is not a constant in her life or actively involved the way my family friends & loves ones are, he does see her occassionally & I have no doubt that he loves Mailyna very much, so I am sorry that everyone who has read it will perceive him in that light, regardless of whether or not the statement is true. So to her dad, I'm sorry.